
Effect of Clinically Used Microtubule Targeting Drugs on Viral Infection and Transport Function

 , , , , , , , , , ,
, , , , , , , , , ,  , , and
, , and
Abstract
:1. Introduction
2. Results
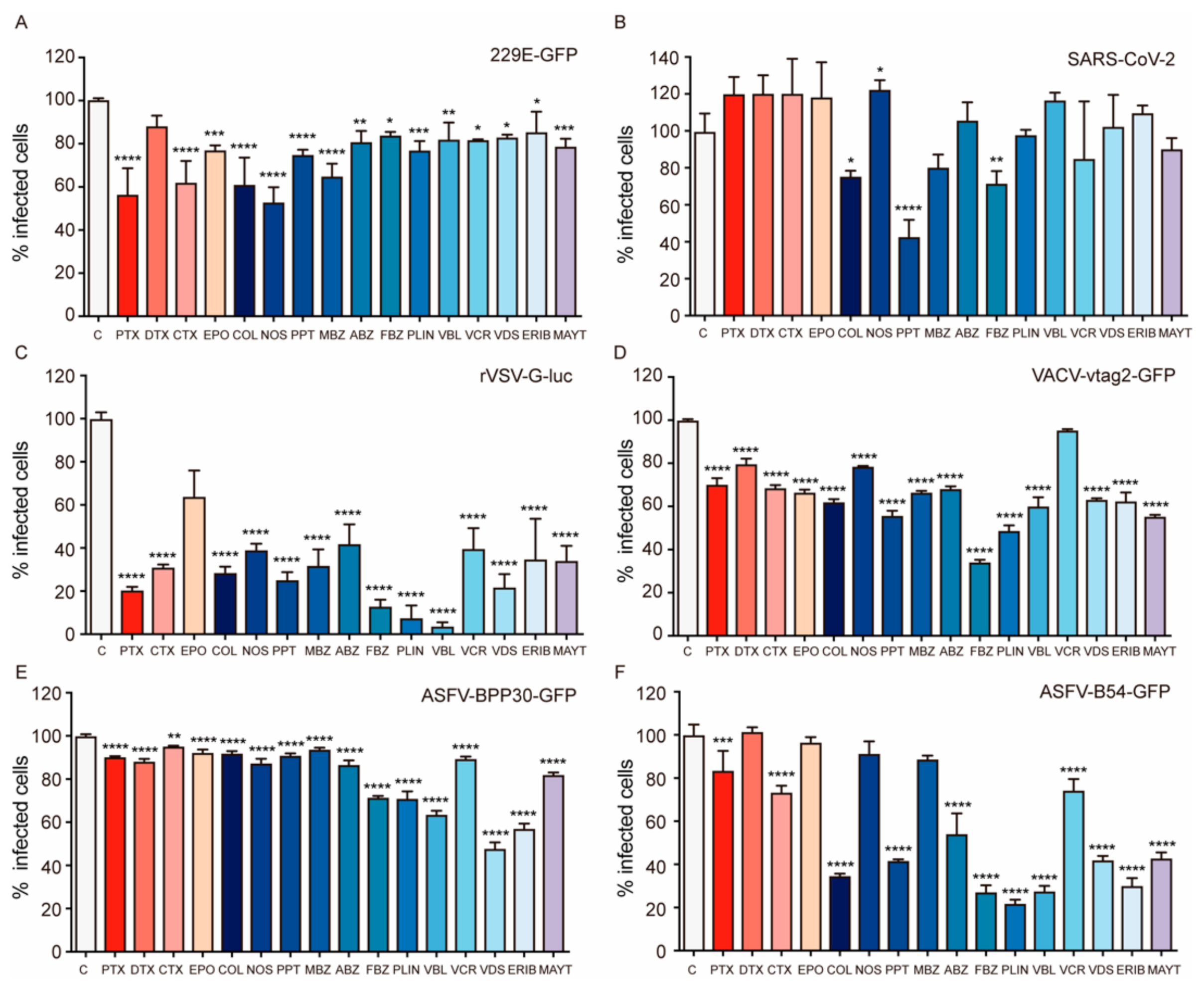
2.1. Effect of MTAs in Viral Infection

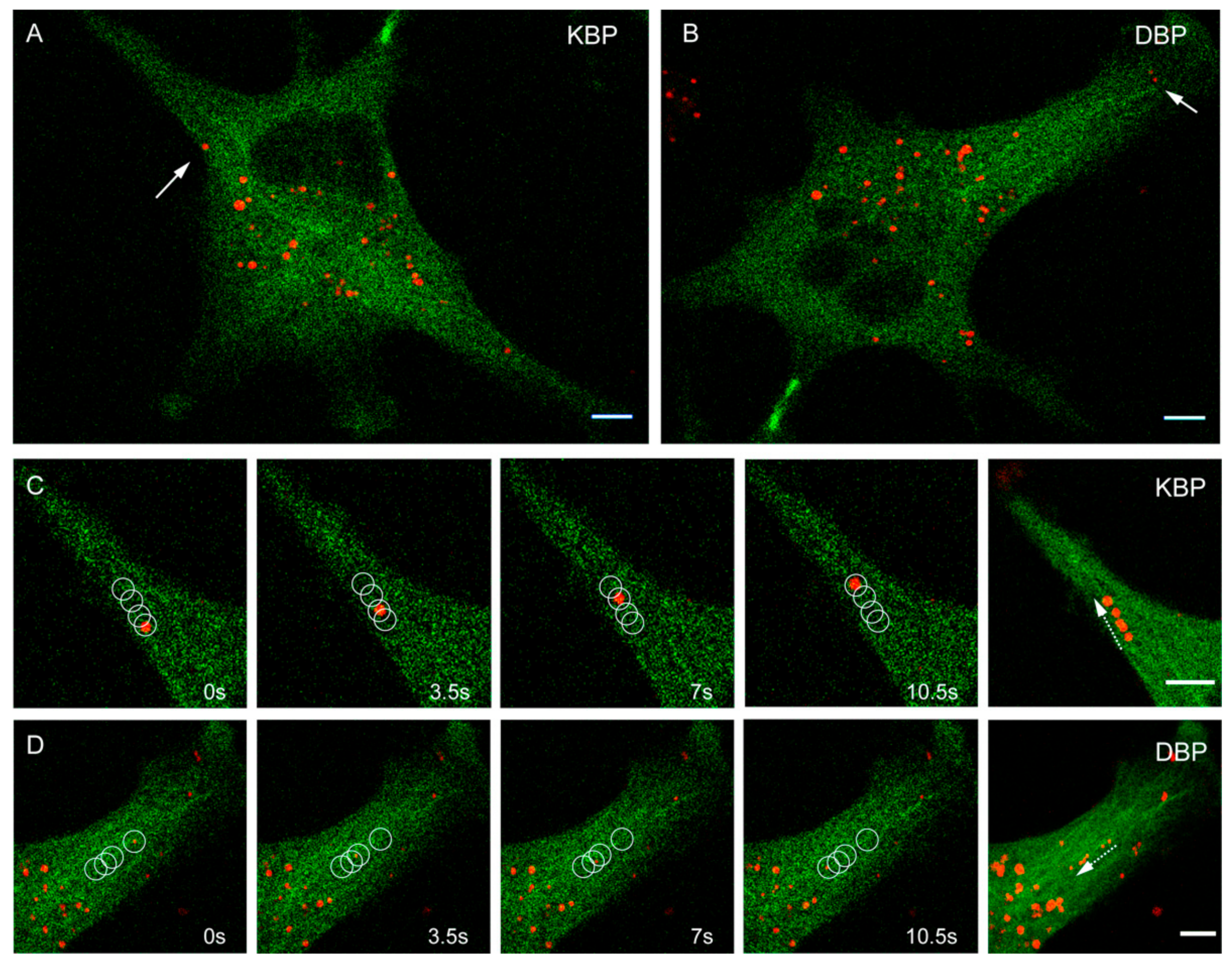
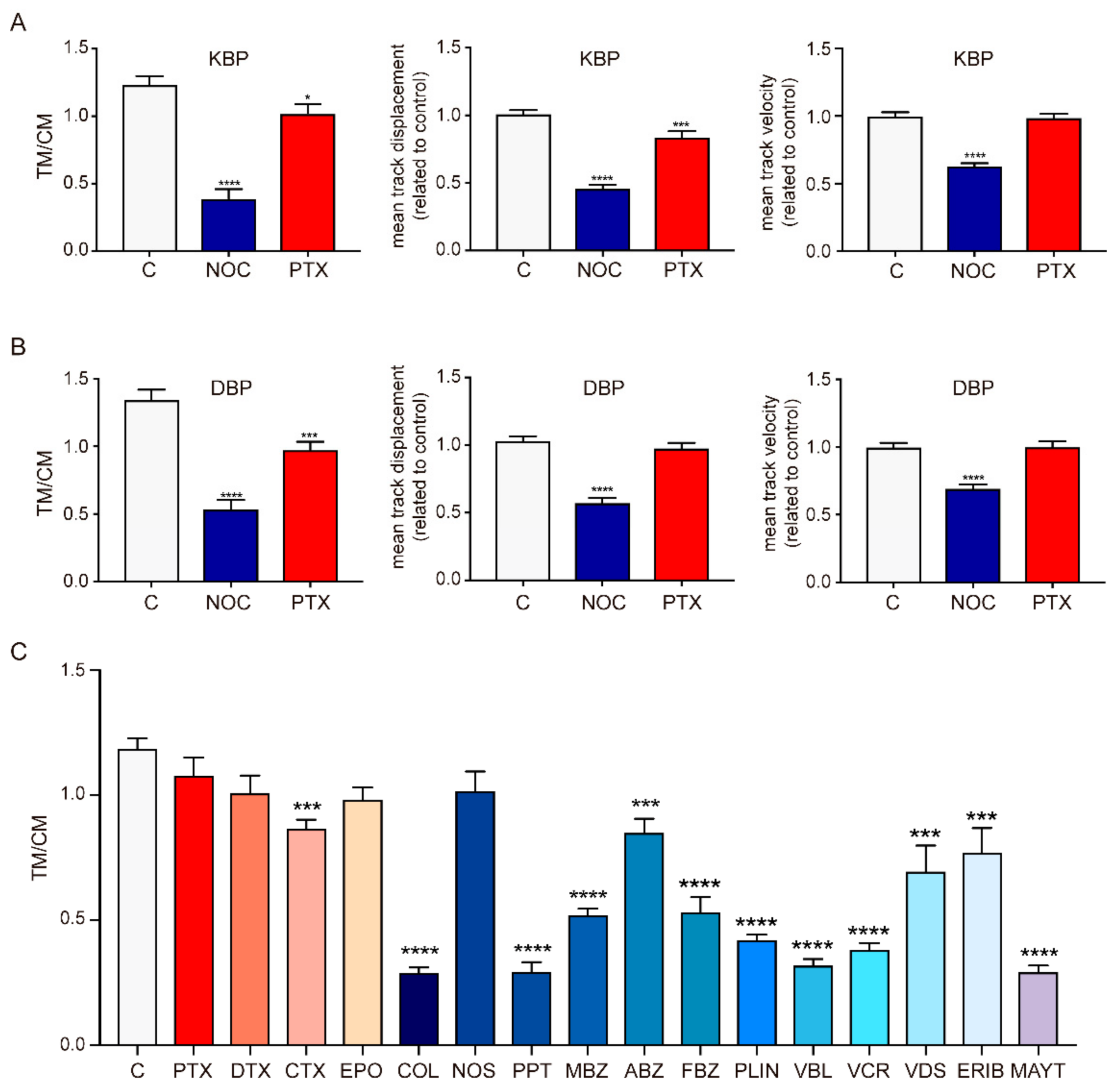
2.2. Effect of MTAs in the Movement of Cellular Motors

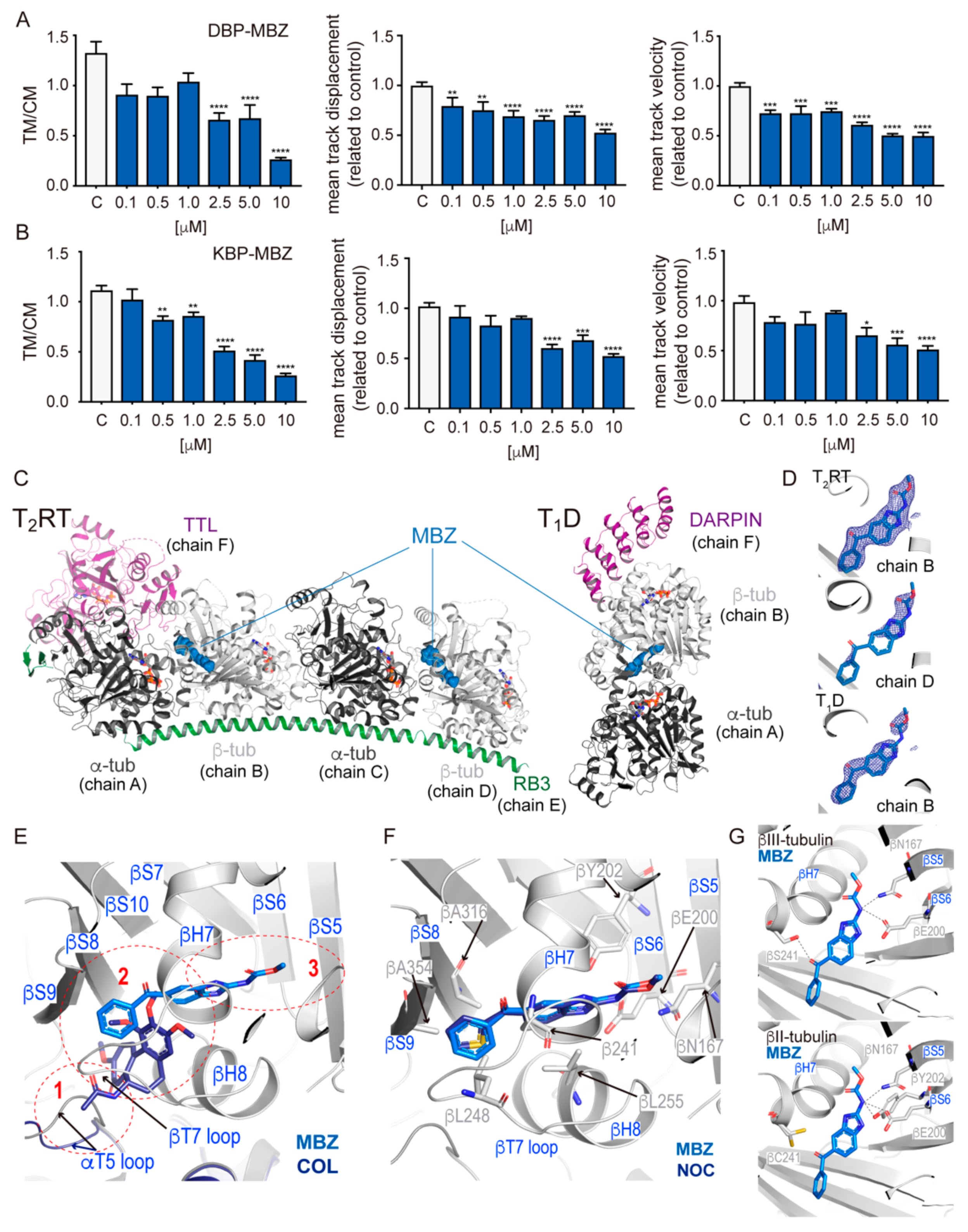
2.3. Biochemical and Structural Characterization of Benzimidazoles Complexes with Mammalian Tubulin
3. Discussion
3.1. MTAs Affect Viral Infections and the Effect Varies According to the Virus Dependency on the Microtubular Network
3.2. MDAs Are More Effective than MSAs in Disrupting Cytosolic Trafficking and Viral Infection
4. Materials and Methods
4.1. Proteins and Chemicals
4.2. Peptide Synthesis and Labeling
4.3. Cell Lines and Cultured Conditions
4.4. Viruses
4.5. In Cell Motility Assays
4.6. Drug Treatment and Cell Viability Determination
4.7. Infection Assays
4.8. Crystallization and Crystal Structure Determination
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saville, M.W.; Lietzau, J.; Pluda, J.M.; Feuerstein, I.; Odom, J.; Wilson, W.H.; Humphrey, R.W.; Feigel, E.; Steinberg, S.M.; Broder, S.; et al. Treatment of HIV-associated Kaposi’s sarcoma with paclitaxel. Lancet 1995, 346, 26–28. [Google Scholar] [CrossRef]
- Slobodnick, A.; Shah, B.; Krasnokutsky, S.; Pillinger, M.H. Update on colchicine, 2017. Rheumatology 2018, 57, i4–i11. [Google Scholar] [CrossRef] [PubMed]
- Messaritakis, J.; Psychou, P.; Nicolaidou, P.; Karpathios, T.; Syriopoulou, B.; Fretzayas, A.; Krikos, F.; Matsaniotis, N. High mebendazole doses in pulmonary and hepatic hydatid disease. Arch. Dis. Child. 1991, 66, 532–533. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, M.O.; Prota, A.E. Microtubule-Targeting Agents: Strategies to Hijack the Cytoskeleton. Trends Cell Biol. 2018, 28, 776–792. [Google Scholar] [CrossRef]
- Matthew, S.; Chen, Q.-Y.; Ratnayake, R.; Fermaintt, C.S.; Lucena-Agell, D.; Bonato, F.; Prota, A.E.; Lim, S.T.; Wang, X.; Díaz, J.F.; et al. Gatorbulin-1, a distinct cyclodepsipeptide chemotype, targets a seventh tubulin pharmacological site. Proc. Natl. Acad. Sci. USA 2021, 118, e2021847118. [Google Scholar] [CrossRef]
- Prota, A.E.; Bargsten, K.; Zurwerra, D.; Field, J.J.; Diaz, J.F.; Altmann, K.H.; Steinmetz, M.O. Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science 2013, 339, 587–590. [Google Scholar] [CrossRef]
- Prota, A.E.; Bargsten, K.; Northcote, P.T.; Marsh, M.; Altmann, K.H.; Miller, J.H.; Díaz, J.F.; Steinmetz, M.O. Structural basis of microtubule stabilization by laulimalide and peloruside A. Angew. Chem. Int. Ed. Engl. 2014, 53, 1621–1625. [Google Scholar] [CrossRef]
- Ravelli, R.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef]
- Gigant, B.; Wang, C.; Ravelli, R.B.; Roussi, F.; Steinmetz, M.O.; Curmi, P.A.; Sobel, A.; Knossow, M. Structural basis for the regulation of tubulin by vinblastine. Nature 2005, 435, 519–522. [Google Scholar] [CrossRef]
- Prota, A.E.; Bargsten, K.; Diaz, J.F.; Marsh, M.; Cuevas, C.; Liniger, M.; Neuhaus, C.; Andreu, J.M.; Altmann, K.-H.; Steinmetz, M.O. A new tubulin-binding site and pharmacophore for microtubule-destabilizing anticancer drugs. Proc. Natl. Acad. Sci. USA 2014, 111, 13817–13821. [Google Scholar] [CrossRef]
- Yang, J.; Wang, Y.; Wang, T.; Jiang, J.; Botting, C.H.; Liu, H.; Chen, Q.; Yang, J.; Naismith, J.; Zhu, X.; et al. Pironetin reacts covalently with cysteine-316 of α-tubulin to destabilize microtubule. Nat. Commun. 2016, 7, 12103. [Google Scholar] [CrossRef]
- Prota, A.E.; Setter, J.; Waight, A.B.; Bargsten, K.; Murga, J.; Diaz, J.F.; Steinmetz, M.O. Pironetin Binds Covalently to alphaCys316 and Perturbs a Major Loop and Helix of alpha-Tubulin to Inhibit Microtubule Formation. J. Mol. Biol. 2016, 428, 2981–2988. [Google Scholar] [CrossRef] [PubMed]
- Vallee, R.B.; Williams, J.C.; Varma, D.; Barnhart, L.E. Dynein: An ancient motor protein involved in multiple modes of transport. J. Neurobiol. 2004, 58, 189–200. [Google Scholar] [CrossRef]
- Verhey, K.J.; Hammond, J.W. Traffic control: Regulation of kinesin motors. Nat. Rev. Mol. Cell Biol. 2009, 10, 765–777. [Google Scholar] [CrossRef]
- Wang, I.-H.; Burckhardt, C.J.; Yakimovich, A.; Greber, U.F. Imaging, Tracking and Computational Analyses of Virus Entry and Egress with the Cytoskeleton. Viruses 2018, 10, 166. [Google Scholar] [CrossRef]
- Döhner, K.; Nagel, C.-H.; Sodeik, B. Viral stop-and-go along microtubules: Taking a ride with dynein and kinesins. Trends Microbiol. 2005, 13, 320–327. [Google Scholar] [CrossRef]
- Ploubidou, A.; Way, M. Viral transport and the cytoskeleton. Curr. Opin. Cell Biol. 2001, 13, 97–105. [Google Scholar] [CrossRef]
- Portilho, D.M.; Persson, R.; Arhel, N. Role of non-motile microtubule-associated proteins in virus trafficking. Biomol. Concepts 2016, 7, 283–292. [Google Scholar] [CrossRef]
- Gornstein, E.; Schwarz, T.L. The paradox of paclitaxel neurotoxicity: Mechanisms and unanswered questions. Neuropharmacology 2014, 76, 175–183. [Google Scholar] [CrossRef]
- Kumar, B.; Kumar, R.; Skvortsova, I.; Kumar, V. Mechanisms of Tubulin Binding Ligands to Target Cancer Cells: Updates on their Therapeutic Potential and Clinical Trials. Curr. Cancer Drug Targets 2017, 17, 357–375. [Google Scholar] [CrossRef]
- Jimenez-Aleman, G.H.; Castro, V.; Londaitsbehere, A.; Gutierrez-Rodríguez, M.; Garaigorta, U.; Solano, R.; Gastaminza, P. SARS-CoV-2 Fears Green: The Chlorophyll Catabolite Pheophorbide A Is a Potent Antiviral. Pharmaceuticals 2021, 14, 1048. [Google Scholar] [CrossRef]
- Ploubidou, A.; Moreau, V.; Ashman, K.; Reckmann, I.; Gonzalez, C.; Way, M. Vaccinia virus infection disrupts microtubule organization and centrosome function. EMBO J. 2000, 19, 3932–3944. [Google Scholar] [CrossRef] [PubMed]
- Ward, B.M.; Moss, B. Vaccinia Virus A36R Membrane Protein Provides a Direct Link between Intracellular Enveloped Virions and the Microtubule Motor Kinesin. J. Virol. 2004, 78, 2486–2493. [Google Scholar] [CrossRef]
- Dodding, M.; Mitter, R.; Humphries, A.C.; Way, M. A kinesin-1 binding motif in vaccinia virus that is widespread throughout the human genome. EMBO J. 2011, 30, 4523–4538. [Google Scholar] [CrossRef]
- Alonso, C.; Miskin, J.; Hernáez, B.; Fernandez-Zapatero, P.; Soto, L.; Cantó, C.; Rodríguez-Crespo, I.; Dixon, L.; Escribano, J.M. African Swine Fever Virus Protein p54 Interacts with the Microtubular Motor Complex through Direct Binding to Light-Chain Dynein. J. Virol. 2001, 75, 9819–9827. [Google Scholar] [CrossRef] [PubMed]
- Hernaez, B.; Díaz-Gil, G.; García-Gallo, M.; Quetglas, J.I.; Rodriguez-Crespo, J.I.; Dixon, L.; Escribano, J.M.; Alonso, C. The African swine fever virus dynein-binding protein p54 induces infected cell apoptosis. FEBS Lett. 2004, 569, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Dalmau-Mena, I.; Del Pino, P.; Pelaz, B.; Cuesta-Geijo, M.; Galindo, I.; Moros, M.; de la Fuente, J.M.; Alonso, C. Nanoparticles engineered to bind cellular motors for efficient delivery. J. Nanobiol. 2018, 16, 33. [Google Scholar] [CrossRef]
- Kawano, T.; Araseki, M.; Araki, Y.; Kinjo, M.; Yamamoto, T.; Suzuki, T. A small peptide sequence is sufficient for initiating kinesin-1 activation through part of TPR region of KLC1. Traffic 2012, 13, 834–848. [Google Scholar] [CrossRef]
- Sirajuddin, M.; Rice, L.M.; Vale, R.D. Regulation of microtubule motors by tubulin isotypes and post-translational modifications. Nat. Cell Biol. 2014, 16, 335–344. [Google Scholar] [CrossRef]
- Shojania Feizabadi, M.; Janakaloti Narayanareddy, B.R.; Vadpey, O.; Jun, Y.; Chapman, D.; Rosenfeld, S.; Gross, S.P. Microtubule C-Terminal Tails Can Change Characteristics of Motor Force Production. Traffic 2015, 16, 1075–1087. [Google Scholar] [CrossRef]
- Ludueña, R.F. Chapter Two—A Hypothesis on the Origin and Evolution of Tubulin. Int. Rev. Cell Mol. Biol. 2013, 302, 41–185. [Google Scholar]
- Vasquez, R.J.; Howell, B.; Yvon, A.M.; Wadsworth, P.; Cassimeris, L. Nanomolar concentrations of nocodazole alter microtubule dynamic instability in vivo and in vitro. Mol. Biol. Cell 1997, 8, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Orr, G.A.; Verdier-Pinard, P.; McDaid, H.; Horwitz, S.B. Mechanisms of Taxol resistance related to microtubules. Oncogene 2003, 22, 7280–7295. [Google Scholar] [CrossRef]
- Rai, A.; Liu, T.; Glauser, S.; Katrukha, E.A.; Estévez-Gallego, J.; Rodríguez-García, R.; Fang, W.-S.; Díaz, J.F.; Steinmetz, M.O.; Altmann, K.-H.; et al. Taxanes convert regions of perturbed microtubule growth into rescue sites. Nat. Mater. 2020, 19, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Alushin, G.M.; Lander, G.C.; Kellogg, E.H.; Zhang, R.; Baker, D.; Nogales, E. High-Resolution Microtubule Structures Reveal the Structural Transitions in αβ-Tubulin upon GTP Hydrolysis. Cell 2014, 157, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Ferro, L.S.; Can, S.; Turner, M.A.; ElShenawy, M.M.; Yildiz, A. Kinesin and dynein use distinct mechanisms to bypass obstacles. eLife 2019, 8, e48629. [Google Scholar] [CrossRef] [PubMed]
- Gard, D.L.; Kirschner, M.W. Microtubule assembly in cytoplasmic extracts of Xenopus oocytes and eggs. J. Cell Biol. 1987, 105, 2191–2201. [Google Scholar] [CrossRef]
- Oliva, M.A.; Prota, A.E.; Rodríguez-Salarichs, J.; Bennani, Y.L.; Jiménez-Barbero, J.; Bargsten, K.; Canales, M.A.; Steinmetz, M.O.; Díaz, J.F. Structural Basis of Noscapine Activation for Tubulin Binding. J. Med. Chem. 2020, 63, 8495–8501. [Google Scholar] [CrossRef]
- Lacey, E. Mode of action of benzimidazoles. Parasitol. Today 1990, 6, 112–115. [Google Scholar] [CrossRef]
- La Regina, G.; Edler, M.C.; Brancale, A.; Kandil, S.; Coluccia, A.; Piscitelli, F.; Hamel, E.; De Martino, G.; Matesanz, R.; Díaz, J.F.; et al. Arylthioindole inhibitors of tubulin polymerization. 3. Biological evaluation, structure-activity relationships and molecular modeling studies. J. Med. Chem. 2007, 50, 2865–2874. [Google Scholar] [CrossRef]
- Xu, K.; Schwarz, P.M.; Ludueña, R.F. Interaction of nocodazole with tubulin isotypes. Drug Dev. Res. 2002, 55, 91–96. [Google Scholar] [CrossRef]
- Pecqueur, L.; Duellberg, C.; Dreier, B.; Jiang, Q.; Wang, C.; Plückthun, A.; Surrey, T.; Gigant, B.; Knossow, M. A designed ankyrin repeat protein selected to bind to tubulin caps the microtubule plus end. Proc. Natl. Acad. Sci. USA 2012, 109, 12011–12016. [Google Scholar] [CrossRef]
- La Sala, G.; Olieric, N.; Sharma, A.; Viti, F.; Balaguer-Perez, F.A.; Huang, L.; Tonra, J.R.; Lloyd, G.K.; Decherchi, S.; Díaz, J.F.; et al. Structure, Thermodynamics, and Kinetics of Plinabulin Binding to two Tubulin Isotypes. Chem 2019, 5, 2969–2986. [Google Scholar] [CrossRef]
- Massarotti, A.; Coluccia, A.; Silvestri, R.; Sorba, G.; Brancale, A. The Tubulin Colchicine Domain: A Molecular Modeling Perspective. ChemMedChem 2012, 7, 33–42. [Google Scholar] [CrossRef]
- Lacey, E.; Prichard, R.K. Interactions of benzimidazoles (BZ) with tubulin from BZ-sensitive and BZ-resistant isolates of Haemonchus contortus. Mol. Biochem. Parasitol. 1986, 19, 171–181. [Google Scholar] [CrossRef]
- Kwa, M.S.G.; Veenstra, J.G.; Van Dijk, M.; Roos, M.H. β-Tubulin Genes from the Parasitic Nematode Haemonchus contortus Modulate Drug Resistance in Caenorhabditis elegans. J. Mol. Biol. 1995, 246, 500–510. [Google Scholar] [CrossRef]
- Ghisi, M.; Kaminsky, R.; Mäser, P. Phenotyping and genotyping of Haemonchus contortus isolates reveals a new putative candidate mutation for benzimidazole resistance in nematodes. Vet. Parasitol. 2007, 144, 313–320. [Google Scholar] [CrossRef]
- Tan, T.K.; Lim, Y.A.L.; Chua, K.H.; Chai, H.C.; Low, V.L.; Bathmanaban, P.; Affendi, S.; Wang, D.; Panchadcharam, C. Characterization of benzimidazole resistance in Haemonchus contortus: Integration of phenotypic, genotypic and proteomic approaches. Parasitol. Res. 2020, 119, 2851–2862. [Google Scholar] [CrossRef]
- Jouvenet, N.; Monaghan, P.; Way, M.; Wileman, T. Transport of African Swine Fever Virus from Assembly Sites to the Plasma Membrane Is Dependent on Microtubules and Conventional Kinesin. J. Virol. 2004, 78, 7990–8001. [Google Scholar] [CrossRef]
- Hernáez, B.; Tarragó, T.; Giralt, E.; Escribano, J.M.; Alonso, C. Small peptide inhibitors disrupt a high affinity interaction between cytoplasmic dynein and a viral cargo protein. J. Virol. 2010, 84, 10792–10801. [Google Scholar] [CrossRef]
- Carter, G.C.; Rodger, G.; Murphy, B.J.; Law, M.; Krauss, O.; Hollinshead, M.; Smith, G.L. Vaccinia virus cores are transported on microtubules. J. Gen. Virol. 2003, 84, 2443–2458. [Google Scholar] [CrossRef]
- Hollinshead, M.; Rodger, G.; Van Eijl, H.; Law, M.; Hollinshead, R.; Vaux, D.; Smith, G.L. Vaccinia virus utilizes microtubules for movement to the cell surface. J. Cell Biol. 2001, 154, 389–402. [Google Scholar] [CrossRef]
- Yacovone, S.K.; Smelser, A.; Macosko, J.C.; Holzwarth, G.; Ornelles, D.A.; Lyles, D.S. Migration of Nucleocapsids in Vesicular Stomatitis Virus-Infected Cells Is Dependent on both Microtubules and Actin Filaments. J. Virol. 2016, 90, 6159–6170. [Google Scholar] [CrossRef] [PubMed]
- Das, S.C.; Nayak, D.; Zhou, Y.; Pattnaik, A.K. Visualization of intracellular transport of vesicular stomatitis virus nucleocapsids in living cells. J. Virol. 2006, 80, 6368–6377. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Zhang, Y.; Lin, Z.; Shi, K.; Jiu, Y. Cytoskeleton—A crucial key in host cell for coronavirus infection. J. Mol. Cell Biol. 2021, 12, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Rüdiger, A.-T.; Mayrhofer, P.; Ma-Lauer, Y.; Pohlentz, G.; Müthing, J.; von Brunn, A.; Schwegmann-Weßels, C. Tubulins interact with porcine and human S proteins of the genus Alphacoronavirus and support successful assembly and release of infectious viral particles. Virology 2016, 497, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Naghavi, M.H.; Walsh, D. Microtubule regulation and function during virus infection. J. Virol. 2017, 91, e00538-17. [Google Scholar] [CrossRef] [PubMed]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14, 454–460. [Google Scholar] [CrossRef]
- Khan, N.; Chen, X.; Geiger, J.D. Role of Endolysosomes in Severe Acute Respiratory Syndrome Coronavirus-2 Infection and Coronavirus Disease 2019 Pathogenesis: Implications for Potential Treatments. Front. Pharmacol. 2020, 11, 595888. [Google Scholar] [CrossRef]
- Bayati, A.; Kumar, R.; Francis, V.; McPherson, P.S. SARS-CoV-2 infects cells after viral entry via clathrin-mediated endocytosis. J. Biol. Chem. 2021, 296, 100306. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Dimitrov, D.S. Virus entry: Molecular mechanisms and biomedical applications. Nat. Rev. Genet. 2004, 2, 109–122. [Google Scholar] [CrossRef]
- Reck-Peterson, S.L.; Redwine, W.B.; Vale, R.D.; Carter, A.P. The cytoplasmic dynein transport machinery and its many cargoes. Nat. Rev. Mol. Cell Biol. 2018, 19, 382–398. [Google Scholar] [CrossRef]
- Harada, A.; Takei, Y.; Kanai, Y.; Tanaka, Y.; Nonaka, S.; Hirokawa, N. Golgi Vesiculation and Lysosome Dispersion in Cells Lacking Cytoplasmic Dynein. J. Cell Biol. 1998, 141, 51–59. [Google Scholar] [CrossRef]
- Granger, E.; McNee, G.; Allan, V.; Woodman, P. The role of the cytoskeleton and molecular motors in endosomal dynamics. Semin. Cell Dev. Biol. 2014, 31, 20–29. [Google Scholar] [CrossRef]
- Cohen, T.; Schwarz, T.; Vigant, F.; Gardner, T.; Hernandez, R.E.; Lee, B.; Tortorella, D. The Microtubule Inhibitor Podofilox Inhibits an Early Entry Step of Human Cytomegalovirus. Viruses 2016, 8, 295. [Google Scholar] [CrossRef]
- Friedman, J.R.; Webster, B.M.; Mastronarde, D.N.; Verhey, K.J.; Voeltz, G.K. ER sliding dynamics and ER-mitochondrial contacts occur on acetylated microtubules. J. Cell Biol. 2010, 190, 363–375. [Google Scholar] [CrossRef]
- Andreu, J.M. Large Scale Purification of Brain Tubulin with the Modified Weisenberg Procedure. In Microtubule Protocols; Zhou, J., Ed.; Humana Press: Totowa, NJ, USA, 2007; pp. 17–28. [Google Scholar]
- Prota, A.E.; Magiera, M.M.; Kuijpers, M.; Bargsten, K.; Frey, D.; Wieser, M.; Jaussi, R.; Hoogenraad, C.C.; Kammerer, R.A.; Janke, C.; et al. Structural basis of tubulin tyrosination by tubulin tyrosine ligase. J. Cell Biol. 2013, 200, 259–270. [Google Scholar] [CrossRef]
- Weinert, T.; Olieric, N.; Cheng, R.; Brünle, S.; James, D.; Ozerov, D.; Gashi, D.; Vera, L.; Marsh, M.; Jaeger, K.; et al. Serial millisecond crystallography for routine room-temperature structure determination at synchrotrons. Nat. Commun. 2017, 8, 542. [Google Scholar] [CrossRef]
- Cervantes-Barragan, L.; Züst, R.; Maier, R.; Sierro, S.; Janda, J.; Levy, F.; Speiser, D.; Romero, P.; Rohrlich, P.-S.; Ludewig, B.; et al. Dendritic cell-specific antigen delivery by coronavirus vaccine vectors induces long-lasting protective antiviral and antitumor immunity. mBiology 2010, 1, e00171-10. [Google Scholar] [CrossRef]
- Whitt, M.A. Generation of VSV pseudotypes using recombinant ΔG-VSV for studies on virus entry, identification of entry inhibitors, and immune responses to vaccines. J. Virol. Methods 2010, 169, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Blasco, R.; Moss, B. Selection of recombinant vaccinia viruses on the basis of plaque formation. Gene 1995, 158, 157–162. [Google Scholar] [CrossRef]
- Barrado-Gil, L.; Galindo, I.; Martínez-Alonso, D.; Viedma, S.; Alonso, C. The ubiquitin-proteasome system is required for African swine fever replication. PLoS ONE 2017, 12, e0189741. [Google Scholar] [CrossRef] [PubMed]
- Hernaez, B.; Escribano, J.M.; Alonso, C. Visualization of the African swine fever virus infection in living cells by incorporation into the virus particle of green fluorescent protein-p54 membrane protein chimera. Virology 2006, 350, 1–14. [Google Scholar] [CrossRef]
- Enjuanes, L.; Carrascosa, A.L.; Moreno, M.A.; Vinuela, E. Titration of African Swine Fever (ASF) Virus. J. Gen. Virol. 1976, 32, 471–477. [Google Scholar] [CrossRef]
- Tarantino, N.; Tinevez, J.-Y.; Crowell, E.F.; Boisson, B.; Henriques, R.; Mhlanga, M.; Agou, F.; Israël, A.; Laplantine, E. TNF and IL-1 exhibit distinct ubiquitin requirements for inducing NEMO–IKK supramolecular structures. J. Cell Biol. 2014, 204, 231–245. [Google Scholar] [CrossRef]
- Tinevez, J.-Y.; Herbert, S. The NEMO Dots Assembly: Single-Particle Tracking and Analysis. In Bioimage Data Analysis Workflows; Miura, K., Sladoje, N., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 67–96. [Google Scholar]
- Qian, H.; Sheetz, M.; Elson, E. Single particle tracking. Analysis of diffusion and flow in two-dimensional systems. Biophys. J. 1991, 60, 910–921. [Google Scholar] [CrossRef]
- Levitz, S.M.; Diamond, R.D. A Rapid Colorimetric Assay of Fungal Viability with the Tetrazolium Salt MTT. J. Infect. Dis. 1985, 152, 938–945. [Google Scholar] [CrossRef]
- Estevez-Gallego, J.; Josa-Prado, F.; Ku, S.; Buey, R.M.; Balaguer, F.A.; Prota, A.E.; Lucena-Agell, D.; Kamma-Lorger, C.; Yagi, T.; Iwamoto, H.; et al. Structural model for differential cap maturation at growing microtubule ends. eLife 2020, 9, e50155. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D 2011, 67, 235–242. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A Comprehensive Python-based System for Macromolecular Structure Solution. Acta Crystallogr. Sect. D 2010, 66, 213–221. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Huh-7 Lunet C3 | Vero E6 | Vero | A549-ACE2 | |
|---|---|---|---|---|
| paclitaxel (PTX) | 5 | 25 | 25 | 0.025 |
| docetaxel (DTX) | 0.03 | 10 | 10 | 0.0125 |
| cabazitaxel (CTX) | 0.01 | 25 | 10 | 0.5 |
| ixabepilone (EPO) | 0.05 | 25 | 25 | 0.0125 |
| colchicine (COL) | 0.2 | 25 | 75 | 0.1 |
| noscapine (NOS) | 100 | 100 | 100 | 50 |
| phodophyllotoxin (PPT) | 0.2 | 50 | 50 | 0.1 |
| mebendazole (MBZ) | 5 | 10 | 10 | 2 |
| albendazole (ABZ) | 1 | 25 | 25 | 2 |
| febendazole (FBZ) | 1 | 50 | 75 | 2 |
| plinabulin (PLIN) | 0.5 | 25 | 25 | 0.002 |
| vinblastine (VBL) | 0.05 | 5 | 25 | 100 |
| vincristine (VCR) | 5 | 1 | 1 | 0.4 |
| vindesine (VDS) | 0.02 | 100 | 100 | 0.006 |
| eribulin (ERIB) | 0.01 | 10 | 100 | 0.0025 |
| maytansine (MAYT) | 0.1 | 25 | 25 | 0.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliva, M.Á.; Tosat-Bitrián, C.; Barrado-Gil, L.; Bonato, F.; Galindo, I.; Garaigorta, U.; Álvarez-Bernad, B.; París-Ogáyar, R.; Lucena-Agell, D.; Giménez-Abián, J.F.; et al. Effect of Clinically Used Microtubule Targeting Drugs on Viral Infection and Transport Function. Int. J. Mol. Sci. 2022, 23, 3448. https://doi.org/10.3390/ijms23073448
Oliva MÁ, Tosat-Bitrián C, Barrado-Gil L, Bonato F, Galindo I, Garaigorta U, Álvarez-Bernad B, París-Ogáyar R, Lucena-Agell D, Giménez-Abián JF, et al. Effect of Clinically Used Microtubule Targeting Drugs on Viral Infection and Transport Function. International Journal of Molecular Sciences. 2022; 23(7):3448. https://doi.org/10.3390/ijms23073448
Chicago/Turabian StyleOliva, María Ángela, Carlota Tosat-Bitrián, Lucía Barrado-Gil, Francesca Bonato, Inmaculada Galindo, Urtzi Garaigorta, Beatriz Álvarez-Bernad, Rebeca París-Ogáyar, Daniel Lucena-Agell, Juan Francisco Giménez-Abián, and et al. 2022. "Effect of Clinically Used Microtubule Targeting Drugs on Viral Infection and Transport Function" International Journal of Molecular Sciences 23, no. 7: 3448. https://doi.org/10.3390/ijms23073448